Uso degli oppiacei nel dolore cronico del paziente non oncologico: razionale e criteri di scelta
Opioids in chronic non cancer pain: rational and selection criteria
Rassegna clinica
Pathos 2011, 18; 1; 2011, June 10
_____________________________________________________
Sergio Mameli, Angela Pili, Giovanni Maria Pisanu,
Maura Carboni
SC Medicina del Dolore, PO Oncologico A. Businco, Cagliari
Elisa Marchi
Scuola Specializzazione Anestesia, Rianimazione e Ter. Antalgica
Università di Cagliari
_______________________________________________________
Riassunto L’efficacia terapeutica e la comparsa di effetti collaterali dei farmaci oppiacei è fortemente condizionata dalle caratteristiche individuali del paziente, del quale è impossibile conoscere le peculiari connotazioni farmacogenomiche che determinano quelle espressioni fenotipiche che renderanno più o meno sensibile il paziente al farmaco somministrato e ne determineranno anche la presenza e l’importanza degli effetti collaterali.
La conoscenza del metabolismo degli oppiacei a livello epatico può contribuire a guidare le scelte terapeutiche verso una maggior personalizzazione delle stesse. Sono da preferire, per ridurre al minimo l'imprevedibilità sia degli effetti favorevoli che di quelli collaterali, i farmaci che risentono meno delle interazioni con altri farmaci e che, a loro volta, possano compromettere l’effi cacia delle molteplici associazioni farmacologiche cui sono sottoposti soprattutto i pazienti più anziani, spesso afflitti da gravi compromissioni d’organo.
Summary The therapeutic efficacy and appearance of side effects of opioid drugs is strongly conditioned by the patient’s individual characteristics. It is impossible to know the patient’s particular pharmacogenomic connotations which determine those phenotypic expressions that make the patient more or less sensitive to the administered drug, and that also determine the presence and significance of side effects. Knowledge of the patient’s hepatic opioid metabolization can aid in the choice of a more personalised therapy. To reduce the unpredictability of favourable effects and side effects, it is preferable to choose drugs
that are less affected by pharmacological interactions, and that can also compromise the efficacy of the many pharmacological associations that patients undergo, especially elderly patients, who often suffer from serious organ impairments.
Parole chiave Dolore cronico non oncologico, metabolizzazione epatica degli oppioidi, interazioni farmacologiche
Key words Chronic nonmalignant pain, hepatic opioid metabolization, pharmacological interactions
Introduzione
Il dolore è un’esperienza complessa e soggettiva con dimensioni multiple, che comporta un enorme impatto sulla qualità di vita, con gravi ripercussioni economiche per il sistema sanitario e sociale. In Italia l’incidenza del dolore cronico riguarda il 23 per cento della popolazione con una prevalenza del dolore non oncologico (80 per cento circa): le artropatie degenerative e infiammatorie hanno un’incidenza del 30 per cento, il dolore neuropatico del 28 per cento, la lombosciatalgia del 27 per cento e il dolore ischemico dell’11 per cento.
Circa 5 milioni di pazienti sono affetti da patologie muscolo-scheletriche associate a dolore cronico e, tra queste, le patologie osteoarticolari sono la causa più frequente di dolore cronico e disabilità fisica. Obiettivi della terapia antalgica in questi pazienti sono il recupero funzionale, il ritorno al lavoro, il miglioramento dei rapporti familiari e sociali, l’uscita dal tunnel della depressione, in altre parole il miglioramento della loro qualità di vita.
Il dolore provoca infatti alterazioni funzionali, situazioni di stress e depressione responsabili dell’abbassamento della soglia al dolore. Le scelte terapeutiche di prima linea includono spesso i FANS. La letteratura è concorde nell’affermare il fallimento della terapia analgesica con i FANS, per caduta dell’efficacia o per gli effetti collaterali.
La dose analgesica dei FANS è inferiore alla dose antinfiammatoria per cui, aumentando il dosaggio, aumenta il rischio di eventi avversi a carico dell’apparato gastrointestinale, cardiovascolare e renale. I fenomeni di cronicizzazione per alterata soglia e per alterata elaborazione del segnale mettono in gioco modifiche tali per cui il substrato biochimico strutturale su cui avvengono non lascia ai FANS alcuna possibilità di intervento. Il dolore cronico mal trattato è responsabile dell’assenteismo sul posto di lavoro, della disoccupazione e dell’alterata qualità di vita. Negli anni si è consolidata la consuetudine di utilizzare gli oppioidi per trattare il dolore da moderato a severo che non ha risposto ai farmaci non oppioidi.
Nonostante gli oppioidi abbiano una maggiore potenza analgesica e una vasta gamma di indicazioni rispetto a tutti gli altri farmaci per il controllo del dolore, rimangono poco usati.
Ci sono ancora molti fattori che costituiscono barriere alla prescrizione degli oppioidi: preoccupazione e scarsa conoscenza della dipendenza, pseudo-dipendenza, tolleranza, rischi di abuso, timore di ripercussioni di tipo legale, possibilità di effetti collaterali.
L’attuale legislazione (Legge 38 del 15 Marzo 2010) facilita la prescrizione e l’utilizzo degli oppiacei, rende obbligatoria la monitorizzazione del dolore nei reparti ospedalieri e la sua registrazione in cartella; impone l’obbligo di mettere in atto tutte le misure necessarie per controllare il dolore. Una corretta diagnosi patogenetica guiderà la scelta terapeutica e quindi la possibilità di avere il massimo della risposta attesa dall’utilizzo di questi farmaci.
Diversi studi randomizzati e controllati dimostrano l’efficacia, la tollerabilità degli oppioidi nei dolori artrosici e in alcuni tipi di dolore neuropaticoconseguente a patologie del sistema nervoso periferico. Questi dati sono estremamente interessanti, ma sono qualitativamente e quantitativamente insufficienti di fronte a una pratica clinica che sta assumendo in alcuni Paesi europei e americani dimensioni enormi (Tabella 1). Molte questioni rimangono aperte sulle indicazioni alla prescrizione di una terapia con oppioidi in pazienti con malattie croniche degenerative a lunga sopravvivenza.
{kind=link}
Sono insufficienti i dati sulla qualità dell’analgesia e delle attività della vita quotidiana, sul recupero psicologico e funzionale di questi ammalati, sui rischi di abuso. Vanno indicati gli effetti sedativi e quelli sulle funzioni cognitive, sul sistema
immunitario, di cui sono note le conseguenze cliniche immediate e a distanza. Gli studi clinici e l’esperienza clinica hanno evidenziato come gli oppiacei non si differenziano molto né per l’efficacia né per l’incidenza di effetti collaterali, criteri che hanno sempre fatto la differenza in farmacologia clinica, ma che per la terapia con oppiacei non rappresentano un criterio di scelta.
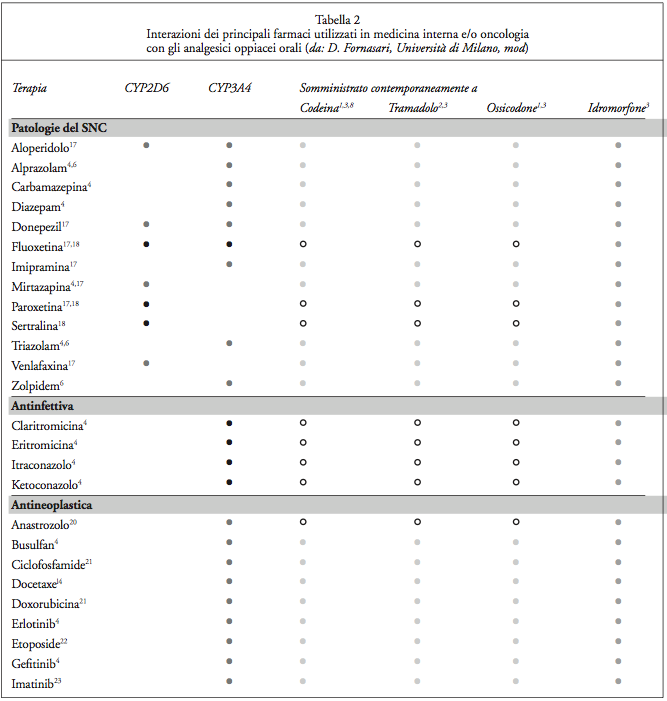
Nella terapia con farmaci oppiacei, invece, rivestono un ruolo determinante la variabilità genetica e le interazioni farmacologiche nel condizionare la risposta attesa, gli eventi avversi e l’interferenza con altri farmaci (Tabella 2,Tabella 2.1, Tabella 2.2).
{kind=link}
{kind=link}
{kind=link}
La variabilità genetica e le interazioni possono avvenire in una qualsiasi delle tappe che caratterizzano il destino di un farmaco dall’assorbimento all’eliminazione ed essere responsabili della risposta individuale agli analgesici oppiacei.
Metabolismo epatico degli oppiacei
L’eliminazione di un farmaco dall’organismo dipende dalla biotrasformazione e dall’escrezione. Ad eccezione del remifentanil, tutti gli oppioidi sono metabolizzati a livello epatico in composti maggiormente idrosolubili poi escreti per via renale e, in misura minore, per via biliare e intestinale. Il metabolismo epatico prevede le classiche reazioni di fase I (ossidazione, riduzione e idrolisi, catalizzate dal sistema del citocromo P-450) e/o di fase II (coniugazione del farmaco o di un suo metabolita con un substrato endogeno come l’acido glicuronico).1
La farmacocinetica analizza la relazione tra dose e concentrazione al sito/i effettore ed è essenzialmente correlata alle proprietà fisico-chimiche della molecola ed ai processi di assorbimento, ridistribuzione, biotrasformazione ed eliminazione del farmaco. La variabilità interindividuale di tali processi rende imprevedibile la cinetica di un dato farmaco in un dato soggetto.
Ciò può spiegare in parte le differenze soggettive osservate nella pratica clinica in termini di efficacia ed effetti avversi. Le reazioni di fase I, in particolar modo, assumono notevole rilevanza in tale contesto. I citocromi P450 sono i principali enzimi catalizzatori delle reazioni di fase I, essi procedono all’ossidazione di sostanze endogene ed esogene in composti maggiormente idrosolubili.2
Si tratta di una superfamiglia di enzimi con almeno 57 geni funzionali noti nella specie umana.3 Al di là del numero elevato di citocromi noti, circa l’80% dei farmaci sono ossidati dagli enzimi 2D6 (30%) e 3A4 (50%). Tali enzimi sono espressi principalmente nel fegato, ma si ritrovano anche nell’intestino tenue (dove possono contribuire a ridurre la biodisponibilità dei farmaci somministrati per os), nei polmoni, nei reni e nella placenta.4
La variabilità nell’attività dei citocromi può influenzare profondamente la risposta ai farmaci in vivo, ciò può tradursi clinicamente in variazioni dell’effi cacia (in eccesso o in difetto) o nello sviluppo di reazioni avverse in seguito a somministrazione della medesima dose standard in soggetti differenti.5,6
Un individuo su quindici può presentare una risposta esagerata o nulla al dosaggio standard di un farmaco. L’attività dei citocromi è in prima istanza geneticamente determinata. Un gene specifico codifica per ogni enzima del CYP450 e ogni soggetto eredita un allele paterno e un allele materno.
Gli alleli sono distinti in “tipo selvaggio” (wild type) o “variante”, con il tipo selvaggio identificato nell’allele che si presenta più comunemente nella popolazione generale. Un “metabolizzatore estensivo” (ovvero, il soggetto con attività enzimatica rientrante nella norma) presenta due coppie di alleli wild type.
Il polimorfismo si verifica quando un allele variante sostituisce uno o entrambi gli alleli wild type. Gli alleli varianti codificano solitamente per un enzima CYP450 che presenta attività ridotta o assente.7 I soggetti con due coppie di alleli varianti sono “poveri metabolizzatori” mentre quelli con un allele “wild type” e uno variante sono classificati come “metabolizzatori intermedi”.
Infine, alcune persone ereditano coppie multiple di alleli “wild type”, il che risulta in un'attività superiore dell’enzima, tale genotipo è definito “metabolizzatore ultrarapido”.8
Bisogna tuttavia tener conto della possibile non corrispondenza tra genotipo e fenotipo, dal momento che tali enzimi possono essere indotti o inibiti nella loro attività da svariati composti. I fenotipi del CYP2D6 metabolizzatori ultrarapidi (UMs), estensivi (EMs), intermedi (IMs) e poveri (PMs), cui corrisponde un decrescendo di attività enzimatica, rappresentano il 3-5 per cento, 70-80 per cento, 10-17 per cento e 5-10 per cento, rispettivamente, della popolazione caucasica.9 Tali percentuali variano tuttavia a seconda dell'etnia considerata, sicché i PMs corrispondono all’1 per cento dei cinesi e al 19 per cento degli afro-americani,10-12 mentre gli UMs rappresentano il 2 per cento dei caucasici svedesi13 e il 16 per cento dei neri etiopi.14
Interazioni farmacologiche
Oltre alla variabile genetica, si deventener presente che la co-somministrazione
di farmaci che seguono la stessa via metabolica conduce inevitabilmente a interazioni tra gli stessi, con possibili effetti deleteri. Molte interazioni farmacologiche registrate nella pratica clinica sono infatti il risultato di un'’alterazione del metabolismo del CYP450.15
I farmaci interagiscono con il sistema CYP450 con modalità differenti. I farmaci possono essere metabolizzati soltanto da un enzima CYP450 o da più enzimi CYP450 contemporaneamente. I farmaci causa di interazioni correlate al metabolismo del CYP450 possono essere distinti in inibitori o induttori. Gli inibitori bloccano l’attività metabolica di uno o più enzimi del CYP450; l’entità con cui un inibitore interessa il metabolismo di un farmaco dipende da fattori quali la dose e la capacità dell’inibitore di legarsi all’enzima. Un farmaco può essere metabolizzato da un enzima e inibirlo allo stesso tempo, o può essere metabolizzato da un enzima e inibirne un altro.
Gli induttori incrementano l’attività dell’enzima CYP450 aumentando la sintesi enzimatica. Differentemente dall’inibizione metabolica, che risulta essere spesso immediata, vi è solitamente un ritardo prima dell’aumento dell’attività dell’enzima, che dipende dall’emivita del farmaco induttore. Un farmaco può inoltre essere metabolizzato dallo stesso enzima CYP450 che induce.
La scoperta di queste interazioni tra geni, ambiente e attività dei citocromi ha portato la FDA ad esigere che il produttore riporti sul foglietto illustrativo informazioni sul metabolismo del farmaco da parte dei CYP450 e sulla sua potenziale capacità di inibizione o induzione degli stessi, per ogni farmaco approvato a partire dal 1997.
Nella pratica clinica è frequente imbattersi in pazienti con dolore cronico affetti da più patologie e sottoposti pertanto a un regime politerapeutico; molti farmaci comunemente prescritti quali antipertensivi, antiaritmici, antidepressivi e gastroprotettori seguono la via del metabolismo epatico attraverso i citocromi, prevalentemente gli isoenzimi 3A4 e 2D6.
La scelta dell’oppioide non può esentarsi in questi casi dalle valutazioni sopra riportate, se si considera in particolar modo che la maggior parte di essi è O-demetilata dagli isoenzimi CYP 2D6 e N-demetilata dai CYP 3A4.
Una rapida panoramica sul metabolismo degli analgesici oppioidi più frequentemente utilizzati, evidenzia come siano poche le molecole disponibili che possano considerarsi esenti dai menzionati fenomeni di variabilità farmacocinetica correlati alla genetica e alle interazioni farmacologiche.
Codeina
La codeina è un analgesico utilizzato in associazione al paracetamolo nel trattamento del dolore lieve, con debole azione agonista sui recettori mu. L’effetto antinocicettivo della codeina è tuttavia principalmente legato alla sua trasformazione in morfina CYP 2D6-mediata.16 Come dimostrato da diversi studi, i PM per il CYP 2D6 sperimentano un effetto analgesico ridotto17 o assente18,19 in seguito ad assunzione di codeina.
Di recente, un case report20 e uno studio caso-controllo21 hanno evidenziato il rischio di depressione neonatale, con conseguenze potenzialmente letali, in caso di assunzione di codeina durante l’allattamento da parte di puerpere con fenotipo UM per il CYP 2D6, per un aumento della concentrazione di morfina nel latte materno.
Ossicodone
L’ossicodone è un oppioide forte semisintetico con metabolismo simile a quello della codeina, che prevede reazioni di O-demetilazione catalizzate principalmente dal CYP 2D6 e di N-demetilazione a opera dei CYP 3A4 e 3A5.
I suoi livelli ematici possono essere incrementati dagli antimicotici (voriconazolo, miconazolo) per inibizione dei citocromi 2D6 e 3A422,23 e ridotti dall’assunzione contemporanea dell’erba di San Giovanni per induzione del CYP 3A4, con riportata riduzione dell’efficacia analgesica riferita dai pazienti.24Anche la rifampicina riduce la concentrazione plasmatica dell’ossicodone orale ed endovenoso, attraverso l’induzione degli enzimi CYP 2D6 e 3A4.25
Metadone
Il metadone è metabolizzato principalmente dal CYP3A4 e in minor misura dal CYP2D6 con produzione di un metabolita inattivo, ed è noto ormai da tempo come le interazioni farmacologiche possano alterarne i livelli in maniera significativa. In generale, i farmaci che inibiscono o inducono il 3A4 (es: rifamicina, carbamazepina, pentobarbital, fenitoina e l’erba di San Giovanni) alterano i livelli sierici di metadone.26 Se da una parte l’induzione del metabolismo può perfino portare allo sviluppo dei sintomi di una sindrome d’astinenza,27 dall’altra l’inibizione del 3A4 da parte di farmaci (es: ciprofloxacina, claritromicina, diltiazem, eritromicina, itraconazolo, ketoconazolo, nefazodone e ritonavir) può condurre rapidamente a tossicità.26Recentemente, l’uso del metadone è stato associato ad un incremento dell’intervallo Qtc ed al rischio di torsioni di punta.28-30 Tale rischio può aumentare con l’uso concomitante di farmaci che ne inibiscono il metabolismo.
Alla luce di tali eventi, nei pazienti complessi dal punto di vista medico, è consigliato attualmente il monitoraggio dei livelli ematici di metadone nella terapia di mantenimento.29
Fentanyl
Il fentanyl è un analgesico popolare ampiamente utilizzato nella sua formulazione transdermica nel trattamento di svariate sindromi dolorose croniche. Anche il fentanyl è principalmente metabolizzato dal 3A4. Il foglietto illustrativo della formulazione transdermica riporta, in neretto, l’avvertenza sul possibile incremento dei livelli ematici in caso di co-somministrazione con inibitori del 3A4 (quali ritonavir, ketoconazolo, itraconazolo, troleandomicina, claritromicina, nelfinavir e nefazodone).31 Il foglietto illustrativo dell’alfentanyl menziona la capacità dell’eritromicina di ridurne significativamente il metabolismo con eventuale sviluppo di depressione respiratoria, ciò è probabilmente dovuto all’inibizione del 3A4 da parte dell’eritromicina.32
È sottolineato inoltre che la cimetidina riduce la clearance dell’alfentanil; il meccanismo è probabilmente simile a quello dell’interazione farmacologica con l’eritromicina, dato che la cimetidina è un inibitore di molti degli enzimi del P450.33. Anche il sufentanyl segue la via metabolica del 3A4.34 Nel più recente foglietto illustrativo non compare alcun riferimento alla farmacocinetica e al metabolismo,35 sebbene siano riportate genericamente le più comuni interazioni farmacologiche. Ad ogni modo è ragionevole considerare che gli inibitori e gli induttori del 3A4 possano alterarne i livelli ematici. È necessaria dunque prudenza qualora tali farmaci vengano somministrati contemporaneamente al sufentanyl.
Tramadolo
Il tramadolo è un composto sintetico simile alla codeina che presenta proprietà
analgesiche uniche. Sembrerebbe essere un profarmaco ed è attivato a livello analgesico dal CYP 450 2D6. L’inibizione del 2D6 da parte di altri farmaci o il deficit genetico del 2D6 possono condurre ad una risposta analgesica effettiva minore. Il tramadolo stesso esercita alcuni effetti analgesici, ma si tratta fondamentalmente di un parziale profarmaco che richiede l’attività del 2D6 (con produzione del metabolita attivo) per un adeguato effetto analgesico. Il 2D6
demetila il tramadolo a un composto chiamato M1, l’isomero destrogiro dell’M1 è associato a una migliore analgesia.36
Poulsen et al37 hanno trovato una risposta analgesica più debole in pazienti PMs per il 2D6. I livelli ematici del M1(+) sono stati dimostrati essere bassi o inesistenti nei poveri metabolizzatori del 2D6, e questi bassi livelli correlano con una peggiore analgesia.38 Inoltre, un disegno di studio a quattro vie sovrapposte placebo-controllato ha dimostrato che l’uso di 20mg di paroxetina (un potente inibitore del 2D6) per 3 giorni riduce in maniera significativa
l’efficacia analgesica di 150mg di tramadolo.39 I livelli di M1(+) sono più bassi quando la paroxetina è usata in combinazione con il tramadolo. Comunque,
la risposta analgesica non è interamente eliminata. Questo può essere dovuto al fatto che il tramadolo stesso ha un debole effetto analgesico. Il tramadolo sembra inoltre essere metabolizzato dal 3 A4 e dal 2D6 attraverso N-demetila-zione.40 Approssimativamente, il 7%-10% della popolazione caucasica è rappresentata da PMs del 2D6.41 Pertanto, un deficit di efficacia analgesica di tramadolo in alcuni individui può essere causato dal fallimento nell’attivazione del farmaco. Inoltre, l’assunzione contemporanea di un potente inibitore del 2D6 può indurre un fenotipo di povero metabolizzatore del 2D6 e può quindi verificarsi se il paziente assume in concomitanza farmaci che inibiscono il 2D6 (bupropione, cimetidina, fluoxetina, metoclopramide, paroxetina, quinidina e ritonavir). In alcuni casi, il deficit di efficacia analgesica da genotipo o fenotipo PMs può essere impropriamente attribuito a un comportamento di craving per il
farmaco da parte dei pazienti. Pertanto, la conoscenza del metabolismo del
tramadolo e del profilo farmacocinetico è imperativa prima della prescrizione
di questo farmaco.
Meperidina e piperidina
La meperidina e altre piperidine sono metabolizzate dal 3A4. Il metabolismo
della meperidina da parte del 3A4 conduce alla formazione di un metabolita tossico, la normeperidina, con una lunga emivita. L’induzione del 3A4 può spingere il sistema a produrre ancora più normeperidina. La meperidina può anche essere associata a un prolungamento dell’intervallo Qtc, proprio come per
il metadone. Queste preoccupazioni hanno condotto alcuni autori a proporre
che la meperidina non venga usata ordinariamente, ma all’occorrenza, dato che esistono analgesici più sicuri, specialmente per l’uso a lungo termine.
Pentazocina e remifentanyl
La pentazocina sembra essere metabolizzata dai CYP 1A2 e dal 3A4.42,43
L’induzione del CYP 1A2 nei fumatori cronici riduce le concentrazioni ematiche di questo farmaco e può condurre ad una riduzione dell’efficacia analgesica. Il remifentanyl non è metabolizzato dal sistema del P450,44 ma si avvale al contrario di un singolare metabolismo organo-indipendente a opera di esterasi tissutali.
Morfina
Anche la morfina è metabolizzata attraverso vie alternative a quelle dei citocromi, essendo per lo più glucuronidata in posizione 3 e 6, con formazione di metaboliti attivi ma idrosolubili, successivamente eliminati per via urinaria.
Idromorfone
L’idromorfone è un analgesico oppiaceo semisintetico che differisce strutturalmente dalla morfina per la sostituzione di un ossigeno al posto del gruppo ossidrilico in posizione 6 e per l’idrogenazione del doppio legame 7-8.45 Benché le stime varino (da 2 a 10 volte), sembra che l’idromorfone assunto per via orale sia circa 5 volte più potente della morfina e abbia una durata d’azione inferiore; è circa 10 volte più liposolubile della morfina.45 A differenza della maggior parte delle molecole oppioidi, è metabolizzato esclusivamente mediante reazioni di fase II, con glucuronidazione a livello epatico e formazione di un metabolita principale privo di attività analgesica, l’idromorfone-3-glucuronide, e di quantità marginali di metaboliti 6-idrossilati.
L’idromorfone, a differenza della morfina, non ha come metabolita il 6-glucuronide (M6G), che presenta attività analgesica e depressiva sul SNC.46
Questo farmaco quindi, non passando attraverso la via dei citocromi per la propria inattivazione ed eliminazione, presenta il grosso vantaggio di eludere l’ampia porzione di variabilità farmacocinetica legata alla genetica ed alle possibili interazioni farmacologiche dei citocromi. Rispetto alla morfina si distingue per l’assenza di metaboliti attivi, e del potenziale rischio di accumulo degli stessi. Presenta una maggior potenza ed una superiore biodisponibilità nella somministrazione orale rispetto alla morfina.
La conoscenza del metabolismo degli oppiacei è di enorme importanza clinica per il medico. Il trattamento individualizzato del dolore comincia con la selezione di un farmaco appropriato.
La scelta può essere complessa quando si è di fronte a pazienti spesso anziani, affetti da altre patologie in terapia polifarmacologica, che può indurre o inibire il sistema del citocromo P450, sottoponendo i metabolizzatori lenti o intermedi a un maggiore rischio.
Nei pazienti sottoposti a trattamenti complessi può essere opportuno iniziare la terapia antalgica con un oppiaceo che non venga metabolizzato dal sistema dei citocromi. Pur non esistendo l’analgesico ideale, occorrerebbe scegliere tra gli oppiacei quello che più si avvicina ai caratteri di quest’ultimo:
• Essere un agonista completo
• Non produrre tolleranza
• Non indurre effetti avversi
• Non avere potenziale di abuso
• Non facilitare iperalgesia
• Avere lunga durata d’azione
• Avere un’alta biodisponibilità orale
• Non essere gravato da importanti interazioni farmacologiche
• Non legarsi in maniera significativa alle proteine plasmatiche
• Non avere metaboliti attivi
• Avere una cinetica lineare
• Essere eliminato per idrolisi in un metabolita non attivo
Questo sarebbe l’analgesico ideale teorico, non ancora realizzato farmacologicamente, ma se su questi parametri confrontiamo gli oppiacei a disposizione, qualcuno si avvicina più di altri a questo profilo.
Published
10th June 2011
Bibliografia
1) Bovril JG. Pharmacokinetics and pharmacodynamics of opioid agonist. Anaesh Pharmacol Rev 1: 122, 1993.
2) Nebert DW, Russel DW. Clinical importance of the cytochromes P450. Lancet 2002; 360: 1155-62.
3) Nelson D. Cytochrome P450 homepage (online). Available from URL: http//dmelson.utmem.edu/CytochromeP450.html1.
4) Slaughter RL, Edwards DJ. Recent advances: the cytochrome P450 enzymes. Ann Pharmacother 1995; 29: 619-624.
5) Ingelman-Sundberg M, Sim SC, Gomez A et al. Infl uence of cytochrome P450 polymorphisms on drug therapies: pharmacogentetics, pharmacoepigenetic and clinical aspects. Pharmacol Ther 2007; 116: 496-526.
6) Zhou SF, Liu JP, Chowbay B. Polymorphism of human cytochrome P450 enzymes and its clinical impact Drug Metab Rev 2009; 41: 89-295.
7) Wilkinson GR. Drug metabolism and variability among patients in drug response. N Engl J Med 2005; 352: 2211-2221.
8) Phillips KA, Veenstra DL, Oren E, Lee JK, Sadee W. Potential role of pharmacogenomics in reducing adverse drug reactions: a systematic review. JAMA 2001; 286: 2270-2279.
9) Sachse C, Brockmoller J, Bauer S et al. Cytochrome P450 2D6 variants in a Caucasian poupolation: allele frequencies and phenotypic consequences. Am J Hum Genet 1997; 60: 284-295.
10) Bradford LD. CYP 2D6 allele frequency in European Caucasian, Asians, Africano and their descendants. Pharmacogenomics 2002; 3: 229-243.
11) Wang SL, Huang JD, Lai MD et al. Molecoular basis of genetic variation in debrisoquin hydroxylation in Chinese subjects: polymorphic in RFLP and DNA sequence of CYp 2D6. Clin Pharmacol Ther 1993; 53: 410-418.
12) Johansson I, Yue QY, Dahl ML et al. Genetic analysis of the interethnic difference between Chinese and Caucasian in the polymorphic metabolism
of debrisoquine and codeine. Eur J Clin Pharmacol 1991; 40: 553-556.
13) Dahl ML, Johansson I, Bertilsson L et al. Ultrarapid Hydroxylation of debrisoquine in Swedish popoulation: analysis of the molecoular genetic
basis. J Pharmacol Exp Ther 1995; 274: 516-520.
14) Akilillu E, Persson I, Bertilsson l et al. Frequent distribution of ultrarapid metabolizer of debrisoquine in an Etiopian population carrying duplicated
and multiduplicated functional CYP 2D6 alleles. J pharmacol Exp Ther 1996; 278: 441-446.
15) Michalets EL. Update: clinically significant cytochrome P- 450 drug interactions. Pharmacotherapy 1998; 18: 84-112.
16) Dayer P, Desmeules J, Leeman T et al. Bioactivation of the narcotic drug codeine in human liver is mediated by the polymorphic monooxygenase
catalyzing debrisoquine 4-hydroxylation (cytochrome P-450 dbl/bufl ). Biochem Biophys Res Commun 1988; 152: 411-416.
17) Lotscj J, Skarke C, Leifhold J et al. Genteic predictors of the clinical response to opioid analgesics: clinical utility and future prespectives. Clin Pharmacokinetic 2004; 43: 983-1013.
18) Eckhardt K, Li S, Ammon S et al. Same incidente of aderse drug events after codeine administration irrespective of the genetically determined differences in morphine formation. Pain 1998; 76: 27-33.
19) Poulsen l, Brosen K, Ardent-Neilsen L et al. Codeine and morphine in extensive and poor metabolizer of sparteine: pharmacokinetics, analgesic effect and side effects. Eur J Clin Pharmacol 1996; 51: 289-295.
20) Koren G, Cairns J, Chitayat D et al. Pharmacogenetics of morphine poisoning in a breastfed neonate of a codeine-prescribed mother. Lancet 2006; 368: 704.
21) Madadi P, Ross CJ, Hayden MR et al. Pharmacogenetics of neonatal opioid toxicity following maternal use of codeine durino brestfeeding: a case-control study. Clin Pharmacol Ther 2009; 85: 31-35.
22) Hagelberg NM, Nieminen TH, Saari TI et al. Interaction of oxycodone and voriconazole-a case series of patients with cancer pain supports the fi ndings of randomised controlled studies with healthy subjects. Eur J Clin Pharmacol. 2011 Jan 11.
23) Grönlund J, Saari TI, Hagelberg N et al. Miconazole oral gel increases the exposure to oral oxycodone by inhibition of CYP2D6 and CYP3A4. Antimicrob Agents Chemother. 2010 Dec 20.
24) Nieminen TH, Hagelberg NM, Saari TI, et al. St John’s wort greatly reduces the concentrations of oral oxycodone. Eur J Pain. 2010 Sep; 14 (8): 854-859.
25) Nieminen TH, Hagelberg NM, Saari TI, et al. Rifampin greatly reduces the plasma concentrations of intravenous and oral oxycodone. Anesthesiology
2009 Jun; 110(6): 1371-1378.
26) Cozza KL, Armstrong SC, Oesterheld JH. Drug Interaction Principles for Medical Practice: 3A4. Washington, DC, American Psychiatric Publishing Inc., 2003, chap. 6, pp 105-107.
27) Lotsch J, Skarke C, Tegeder I et al. Drug interactions with patient-controlled analgesia. Clin Pharmacokinet 2002; 41 :31-57.
28) (No authors listed): Torsades de pointes with methadone. Prescrire Int 2005; 14(76):61-62.
29) Justo D, Gal-Oz A, Paran Y et al. Methadone-associated torsades de pointes (polymorphic ventricular tachycardia) in opioid-dependent patients. Addiction 2006, 101:1333-1338.
30) Ehret GB, Voide C, Gex-Fabry M et al. Drug-induced long-QTsyndrome in injection drug users receiving methadone: high frequency in hospitalized patients and risk factors. Arch Intem Med 2006; 166: 1280-1287.
31) Duragesic package insert, Janssen Pharmaceutica Products, LP, Titusville, NJ, 2003.
32) Alfenta package insert, Taylor Pharmaceuticals, Decatur, IL, July, 1998.
33) Rendic S. Drug interactions of H2-receptor antagonists involving cytochrome P450 (CYPs) enzymes: from the laboratory to the clinic. Croat Med J 1999; 40:357-367.
34) Tateishi T, Krivomk Y, Ueng Y et al. Identification of human liver cytochrome P450 3A4 as the enzyme responsible for fentanyl and sufentanil N-dealkylation. Anesth Analg 1996; 82:167-172.
35) Sufenta* package insert, Taylor Pharmaceuticals, Decatur, IL, Aprii, 2006.
36) Sindmp SH, Madsen C, Brosen K et al. The effect of tramadol in painful polyneuropathy in relation to serum drug and metabolite levels. Clin Pharmacol Ther 1999; 66: 636-641.
37) Poulsen L, Arendt-Nielsen L, Brosen K et al. The hypoalgesic effect of tramadoi in relation to CYP2D6. Clin Phannacol Ther 1996; 60: 634-644.
38) Enggaard TP, Poulsen L, Arendt-Nielsen L et al. The analgesic effect of tramadol after intravenous injection in healthy volunteers in relation to CYP2D6. Anesth Analg 2006; 102: 146-150.
39) Laugesen S, Enggaard TP, Pedersen RS et al. Paroxetine, a cytochrome P450 2D6 inhibitor, diminishes the stereoselective 0 - demethylation and reduces the hypoalgesic effect of tramadol. Clin Phararncol Ther 2005; 77: 312-323.
40) Ultram ER* package insert, Biovail Corporation, Mississauga.ON LSN 8M5, Canada, December, 2006.
41) Kroemer HK, Eichelbaum M. “It’s the genes, Stupid:” molecular basis and clinica1 consequences of genetic cytochrome P450 2D6 polymorphism. Life Sci 1995; 56:2285-2298.
42) Miller LG. Cigarettes and drug therapy: pharmacokinetic and pharmacodynamic considerations. Clin Pharm 1990; 9: 125-135.
43) Zevin S, Benowitz NL. Drug considerations with tobacco smoking: an update. Clin Pharmacokinet 1999; 36:425-438.
44) Ultiva package insert, Glaxo Wellcome, Kalamazoo, MI, October, 1999.
45) Sarhill N et al. Hydromorphone: pharmacology and clinical applications in cancer patients, Support Care Cancer 2001 Mar; 9 (2): 84-96.
46) Dean M. Opioids in renal failure and dialysis patients. J Pain Symptom Manage 2004: 28:497-504.