Pathogenetic role of the pain threshold
Hypothetical therapeutic perspectives based on the use of drugs to elevate it
Hypothetical therapeutic perspectives
based on the use of drugs to elevate it
Review
Pathos 2023; 30. 4. Online 2023, Dec 27_____________________________________________________________________________
Pathos 2023; 30. 4. Online 2023, Dec 27
_____________________________________________________________________________
Guido Orlandini
Pain Medicine
Villa Ravenna, Chiavari-Ge (Italy)
Istituto SYNLAB, Monza-MB (Italy)
_____________________________________________________________________________
Summary
The pain threshold determines the extent of pain perception, and in some cases, its manifestation. Therefore, it is important in certain situations to increase the pain threshold. Measuring the pain threshold is challenging due to its approximate nature. The threshold depends on the number and function of Nav1.7 sodium channels and nerve growth factor (NGF). Unfortunately, we lack therapeutic tools to selectively act on these sodium channels. The clinical use of tanezumab, a drug that can counteract NGF, has not been approved. It is potentially dangerous to misuse drugs that can increase the pain threshold as it may divert attention from treating the underlying cause of pain or even dangerously limit its protective function. However, they can be particularly useful in pathologies where the main pathogenetic mechanism is a reduced pain threshold, such as migraine, musculotensive headache, trigeminal neuropathy, phantom limb pain, fibromyalgia, and others. They are also helpful in situations where pain is no longer useful for diagnostic purposes and when it is due to an incurable condition like cancer.
Riassunto
La soglia del dolore determina l’entità della percezione soggettiva del dolore e in alcuni casi addirittura la sua comparsa. Da questo deriva, sul piano teorico, l’importanza in determinate situazioni di innalzare la soglia del dolore. Purtroppo, la soglia del dolore può essere misurata soltanto molto approssimativamente e, pur sapendo che dipende almeno in parte dal numero e dalla funzionalità dei canali del sodio Nav1.7 e del nerve growth factor (NGF), non si hanno gli strumenti terapeutici per agire selettivamente su quei canali del sodio e si è dovuto rinunciare al farmaco in grado di contrastare il NGF (tanezumab) perchè il suo impiego clinico non è stato approvato. Non c’è dubbio che sarebbe alquanto pericoloso l’abuso dei farmaci in grado di elevare la soglia del dolore perché potrebbe addirittura stornare l’attenzione dalla terapia della causa del dolore o limitarne pericolosamente la funzione protettiva. Tuttavia, in mani esperte essi sarebbero utilissimi nelle patologie dove il principale meccanismo patogenetico è proprio la ridotta soglia del dolore (emicrania, cefalea muscolotensiva, neuropatia trigeminale, dolore dell’arto fantasma, fibromialgia e molte altre), nelle situazioni dove si può ritenere che la soglia del dolore condizioni la gravità della sintomatologia, quando il dolore non è più utile ai fini diagnostici e quando è dovuto a una patologia che non può essere curata come il cancro.
Key words
Pain, threshold. canalopathy, Nav1.7, NGF, tanezumab
Pain, threshold. canalopathy, Nav1.7, NGF, tanezumab
Parole chiave
Dolore, soglia, canalopatia, Nav1.7, NGF, tanezumab
Introduction
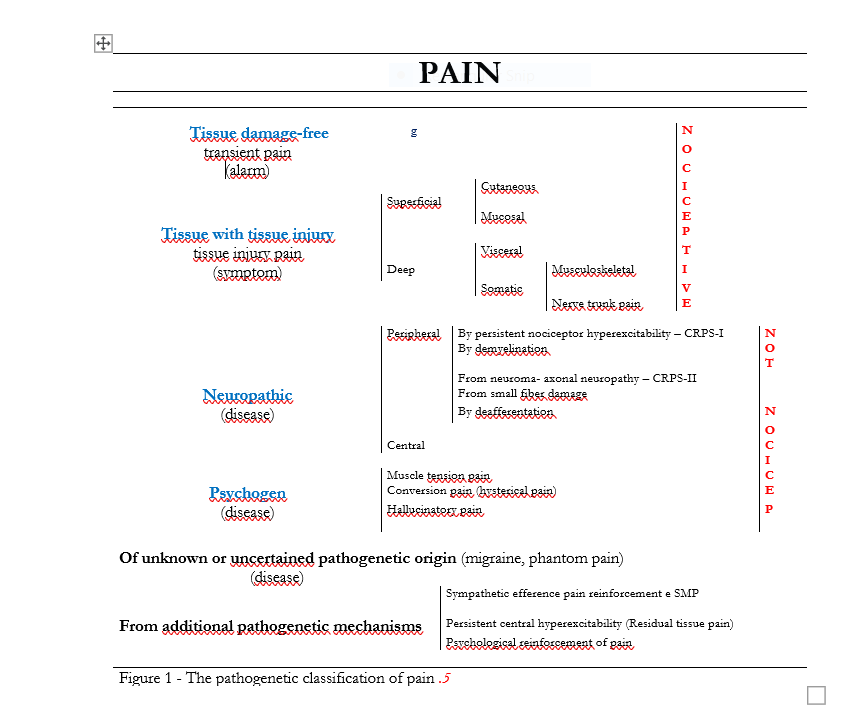
The pathophysiology of pain teaches that various pathogenetic mechanisms activated by different tissue or nerve injuries cause pain (Figure 1). Some observations, however, suggest that an additional concept should be added to this knowledge: I refer to the role of pain threshold, both to explain the occurrence or absence of pain in the presence of the same algogenic lesion,1-3, as well as in explaining its different intensity in patients with apparently the same pathologies, and finally in relation to the production of some pathologies that have always been ambiguously considered to be of unknown or uncertain origin such as Migraine, Phantom Limb Pain, Vaginismus,4 and the much fashionable as inconsistent Fibromyalgia.
Definition of "pain threshold"
Before starting this discussion, although it seems obvious, the definition of "pain threshold" deserves clarification because in reviewing the literature on the subject there is no shortage of confusion. When it is not even confused with hyperalgesia and allodynia .6-13 or with pain tolerance, pain threshold often means its level obtained following the administration of an analgesic,14 infiltrative or surgical treatment15 and other.16-18 For example, NSAIDs that act by blocking the synthesis of prostaglandins, which in turn increase the excitability of nociceptors, elevate the pain threshold because they reduce the sensitizing effect of prostaglandins on nociceptors and thus in addition to controlling the one already present, theoretically can also limit at least some of the pain caused by a subsequently applied painful stimulus. More so, local anesthetics that, by blocking sodium channels, prevent activation of nociceptors control pain and elevate the pain threshold by preventing that caused by a subsequent stimulus. Opioids that reduce synaptic transmission of the nociceptive message between the first and second neurons elevate the pain threshold because in addition to controlling pain they can prevent or at least limit pain caused by a subsequent painful stimulus. In fact, all these treatments modify by elevating the pain threshold, however, it is not the value thus obtained that we are interested in relation to the production of pain due to the various pathologies, but rather the so-called "basal" value in a given individual (thus clearly "subjective"), related to the particular biological substrate at the molecular level and perhaps to the degree of central excitability. Finally, by "drugs to elevate the pain threshold" we must specifically mean those capable of elevating the latter. It seems that these considerations take for granted what the pain threshold is: however, although the concept is apparently intuitive, no clear definition of it emerges from them.
On the other hand, the definition proposed by IASP19 is questionable and useless for our purposes. In fact, IASP defines pain threshold as "...the minimum experience of pain that a subject is able to recognize..." and rejects the definition "...the minimum stimulus intensity at which a subject perceives pain..." because it refers to a stimulus and not to the subjective experience of pain. In fact, even when (in accordance with IASP) we define allodynia as "...pain caused by a stimulus that does not normally cause pain...," we are referring to the stimulus and not to the subjective experience of pain.
The diction "...minimal experience of pain that a subject is able to recognize..." refers only to the subjective experience of pain, and this aspect, which IASP considers a merit, is actually a limitation. In contrast, the phrase "...minimum stimulus intensity at which a subject perceives pain..." by including a factor (the stimulus) that contributes to the production of pain lends clinical utility to the concept of pain threshold.
Since there is no reason to dissociate the stimulus from the pain experience that is a consequence of it, we could say that the pain threshold is "the intensity of the nociceptive stimulus capable of causing pain." This definition is unexceptionable when referring to nociceptive pain but not when it is produced by non-nociceptive stimuli. The fact that in the presence of the same nerve damage some subjects express neuropathic pain and others do not, seems intuitively correlated with a different pain threshold but in this case nociception is not at stake.
Thus, referring back to the definition of pain proposed by IASP,19 which speaks of "unpleasant sensory and emotional experience associated with actual or potential tissue damage or described in terms of such damage" (...or, according to recent and unnecessary updates, "resembling such damage"), the more correct definition of pain threshold might be: "the extent of damage capable of causing pain" which allows it to refer to both nociceptive and neuropathic pain. Moreover, if we use the term "stimulus" we are referring to something that is produced by the damage and that activates the nociceptors, if on the other hand we use the term "damage" we do not necessarily include the stimulus of the nociceptors and therefore we can extend the concept to non-nociceptive pain such as neuropathic pain.
To close this topic, which is in danger of becoming monotonously inconclusive, and to express a personal opinion, I believe that pain threshold should be understood as "that which determines the different subjective response to the same nociceptive or dysnociceptive stimulus."
Finally, anticipating the objection, a separate case is psychogenic pain where one can hardly think of a threshold.
Pathogenetic role of pain threshold
It is possible that in some cases in order to actually cause pain or more generally to cause more or less severe pain, the classical pathogenetic mechanisms of pain (Figure 1) require a particular biological substrate at the molecular level that determines the value of the pain threshold, likely governed by the function of Nav1.7 sodium channels, of nerve growth factor (NGF), and possibly many other factors such as involvement of TRP-channels, activation of microglia in the dorsal horn of the spinal cord,20 activation of Cav3.2-T-type21 and probably others whose existence we still ignore.
{kind=link}
It is normal that anatomical damage of a certain magnitude (e.g., a bone fracture), with the aim of protecting the organism, activates the nociceptive signal and produces more or less intense pain depending on the severity of the damage and thus on the magnitude of the nociceptive message, which in turn depends on the amount of receptors and sensory fibers involved and, backwards, on the amount of activated Nav1.7 sodium channels.
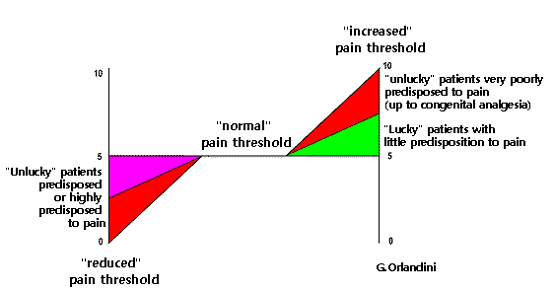
Under normal conditions, one can therefore assume a proportion between the magnitude of nociception-dysnociception and pain intensity that corresponds to a "normal pain perception threshold": however, if due to dysfunction of Nav1.7 sodium channels ("channelopathy") this threshold is altered, in the presence of a nociceptive-dysnociceptive stimulus of equal severity, pain may be felt with greater or lesser intensity or not at all. Thus we have that for a mild nociceptive-dysnociceptive stimulus some individuals experience particularly severe pain and others little or no pain (Figure 2).
{kind=link}
Channelopathy explains why the same anatomical damage to peripheral nerves causes pain in some individuals and not in others and underlies painful conditions such as Neuropathic Pain from Small Fiber Pathology, Burning Mouth Syndrome, Trigeminal Neuropathy due to peripheral nerve damage (to produce which a simple tooth extraction is sufficient) that mostly goes clinically unnoticed but in some cases causes severe pain, Migraine (where the only anatomical finding and classically considered the cause of pain is an intracranial vasodilatation that is completely harmless in most people but a source of pain in migraineurs), Musculotensive Headache (where there is only the anatomical finding of prolonged contraction of the epicranial muscles), Phantom Limb Pain, Fibromyalgia (which is nothing more than the sum of a series of relatively modest tissue damage), Irritable Bowel Syndrome (typically without a clear organic basis), and, finally, common situations such as Systemic Degenerative Disease Pain in the elderly.22
Considering channelopathy by no means means that one should not investigate the pathogenetic mechanisms of pain in order to choose the appropriate therapy to counteract them,23 but how effective the treatment will be might depend on the pain threshold so it would be appropriate to act on the mechanism of production of nociception or dysnociception and at the same time try to raise the pain threshold if it is particularly low.
Role of Nav1.7 sodium channels in nociceptive pain
The importance of Nav1.7 sodium channels in the production of nociceptive pain is demonstrated by the observation of Kwon et al.24 who showed that Nav1.7 sodium channels increase following the provocation of inflammatory pulpitis.
Role of Nav1.7 sodium channels in neuropathic pain
The role of Nav1.7 sodium channels in neuropathic pain had already been reported by the observation that a genetic mutation that increases their activity is responsible for Erythromelalgia,25 by the finding that they increase in nerve damage26 contributing to its production, and that hydrogen sulfide that increases their activity exacerbates neuropathic pain.27
Neuropathic pain from persistent nociceptor hyperexcitability and small fiber damage
Estacion et al.28 argued that hyperfunction of Nav1.7 sodium channels is responsible not only for hereditary Erythromelalgia (characterized by burning pain felt in the distal extremities triggered by heat and relieved by cold), but also for Idiopathic rectal hypersensitivity syndrome and extreme paroxysmal pain and Neuropathic pain from small fiber damage.29 In this regard, the same Authors reported that increased Nav1.7 sodium channel function is present in approximately 30% of patients with positive biopsy for small fiber pathology, and this was later confirmed by others.30-31
Pain in CRPS-I
Almost incomprehensible is the observation that Nav1.7 sodium channels are not implicated in CRPS-I;32 this statement (which as far as I could verify is not confirmed in the literature) is rather strange if we consider as the primum movens of CRPS-I the hyperexcitability of nociceptors implying a prominent role of Nav1.7 sodium channel hyperfunction.
Neuropathic pain from axonal neuropathy
In neuropathic pain from axonal neuropathy, Nav1.7 sodium channels play an important role in producing depolarization in neuroma neonoreceptors and in producing depolarization of the second neuron with sodium entry through post-synaptic membrane ion channels by transferring to it the message conducted by Aβ fibers proliferated to replace C fibers as a result of morphofunctional remodeling of the DREZ. On the other hand, to think that these receptors are not present in myelinated fibers I do not think is correct because a role of these receptors is crucial in demyelination pain involving Aβ fibers.
Demyelination neuropathic pain
If we consider that demyelination pain depends on the compensatory incorporation of newly formed sodium channels into demyelinated Aβ fiber tracts and thus their increased numbers, which justifies the efficacy of generically sodium channel inhibitor drugs such as local anesthetics, carbamazepine, and lamotrigine, we must conclude that Nav1.7 sodium channels are heavily implicated in the pathogenesis of this type of neuropathic pain.
Neuropathic deafferentation pain and central pain
The deafferentation pain caused by the nerve injury proximal to the dorsal root ganglion is due to the denervation hyperexcitability of the second neuron and its consequent spontaneous epileptiform discharge: it is difficult to imagine that sodium channels are not involved in this spontaneous activation (depolarization), and the same is true for central pain.
Measurement of pain threshold
To determine pain threshold, one could hypothesize measuring the number or assessing the functionality of Nav1.7 sodium channels, but we do not have the means to implement this type of detection. An indirect method, could be the study of genes that regulate sodium channel trophism, and the maximum proposed in this regard is the assay in serum of the gene that increases the number of sodium channels: the Foxo 1 protein.33
More simply but also much more grossly, one can think of physical semeiological methods such as the simple application of ice34 and others that presuppose the quantification of the mechanical energy required to evoke pain, for example, with acupressure or with the equipment proposed for this purpose that, pompously called algometers, consists of a blunt-tipped piston to be pressed on the part to be examined, connected to a pressure gauge that indicates the pressure we must exert to provoke pain.35-38 This measurement indicates the subjective response in the area where the pressure is applied and thus the pain threshold at that point. It should be reiterated that to get information about an individual's pain threshold, this measurement should not be made in the painful area (where what is detected is allodynia) but on an area where the patient has no spontaneous pain, nor allodynia.39-40 In any case, the most we can get is a general suspicion of a normal, reduced, or increased pain threshold.
Leaving aside the gross method involving the application of electrical stimuli,41 a more refined method might be the use of the old neurometry relying on the fact that with different frequencies of electrical stimulation different categories of nerve fibers are stimulated: in particular, with the 2000 Hz current the Aß fibers are stimulated, with the 250 Hz current the Aδ fibers, and with the 5 Hz current the C fibers. Created to quantitatively assess the sensitivity of a skin territory and especially to highlight and typify allodynia, the instrument used not on the pain area but on areas where the patient has no pain could give information on the excitability of Aβ, Aδ and C fibers.
Means to control pain threshold
Unfortunately, we currently have no means of raising the basal pain threshold by counteracting Nav1.7 sodium channels and NGF!
In the last 20 years and especially in the last 5, new interesting therapeutic targers for the treatment of pain represented by the aforementioned Nav1.7 sodium channels as well as NGF and according to others,42 also by Nav1.8 sodium channels, TRPV1, nitric oxide, prostaglandin E and interleukin-6 have been identified or at least given more serious consideration. To date, among these hypothetical drugs, only one product is available in Italy (and only for veterinary medicine) that is called Bedinvetmab and is analogous to Tanezumab (produced by Zoetis Italia under the trade name Librala and available in vials for subcutaneous use of 5-10-15-20-30 mg to be administered at a dose of 0.5-1 mg/Kg/4 weeks).
There is no shortage of obstructionism toward drugs that counteract Nav1.7 sodium channels and NGF to raise the pain threshold, both trivially because they might be considered commercially competitive against NSAIDs and opioids and because their "dangerousness" cannot be ruled out if they were really effective and used indiscriminately. In fact, a drug that "makes pain feel less" could be a panacea that overshadows the need to investigate the pathogenesis of pain and choose the most appropriate therapies, and in the hands of the growing number of those in the rampant triumph of mediocrity who consider it unnecessarily laborious to think about and correlate concepts when these can be schematized in short summaries easily found "on the net," it could become an even dangerous weapon inducing even the neglect of causal therapy. At this point, any health care provider regardless of his training (if not the patient himself "educated" by the rampant television advertising of health care devices) could manage pain.
Counteracting sodium channels Nav1.7
Current antidepolarizing drugs that act on sodium channels (local anesthetics, carbamazepine, and other antiepileptics) block all types of sodium channels while those implicated in activation of the transmembrane action potential (TAP) in nociceptors by nociceptive stimuli are only sodium channels Nav1.7 and perhaps Nav1.8 and Nav1.9.43-46 Among the other sodium channels, Nav1.1, Nav1.2 and Nav1.6 are involved in neurotransmission in the CNS and by their hyperfunction in the production of epilepsy, Nav1.4 are involved in muscle contractility and Nav1.5 in cardiac activity.47
Involving all types of sodium channels, the current antidepolarizers drugs acting on them can be used as analgesics in tissue pain only with local administration (as local anesthetics) because with systemic administration achieving their sufficient concentration near nociceptors requires unacceptable doses from the point of view of side effects due to the involvement of sodium channels present in other tissues (especially in the heart). In the case of neuropathic pain, the same drugs can be used systemically in situations where there is excessive proliferation of sodium channels as in the demyelinated tracts of Aβ fibers responsible for the demyelination pain of Trigeminal Neuralgia. In this respect, despite some enthusiasm, nothing has been added since the advent of lacosamide.48-56
On the basis of these considerations, drugs that can selectively counteract Nav1.7 sodium channels could really be a revolutionary pain therapy while avoiding side effects and become a viable alternative not (as one would think) to NSAIDs because they do not exert the anti-inflammatory effect but to other analgesics (including opioids) and antiepileptics used in addition to the treatment of epilepsy also as therapy of some neuropathic pain.
The key role that Nav1.7 sodium channels play in the production of pain is proven by the existence of congenital analgesia believed to be due to the hypofunction of these channels.57-60 and by the increase or decrease in pain caused by chemicals that experimentally increase or decrease their function.
It should be noted that likely the reduced function of Nav1.7 sodium channels in addition to reducing pain also causes hyperfunction (probably compensatory) of the endogenous opioid system since naloxone reduces pain control caused by the loss of their function.61
Regarding their anatomical location, Nav1.7 sodium channels would be found not only in the receptor portion of the first sensory neuron (along with the sodium channels called Nad) where they induce TAP production, but also along the axon contributing to its propagation,62 and according to some28-63-64 also in the dorsal root ganglia, cranial nerve ganglia, and sympathetic ganglia. While the role of Nav1. 7 at the level of nociceptors (where they produce depolarization with sodium entering the nerve cell through their respective channels), in the nerve fiber pathway (where in tissue pain they allow the propagation of depolarization to the central terminal of the first neuron, in axonal damage pain they promote depolarization in the neoreceptors of the neuroma with the entry of sodium through their ion channels and in demyelination neuropathic pain they promote depolarization of the axon with the entry of sodium through the newly formed ion channels of the demyelinated tracts of the fiber) and in the synapse (where they promote depolarization of the second neuron with the entry of sodium through the ion channels of the post synaptic and thus the transfer to it of the nociceptive message) is not what they should have in the spinal and cranial ganglia. In fact, the sympathetic ganglia contain the synapse between the pre- and post-ganglionic neuron, but the spinal and cranial ganglia contain only the pyrenophores from which the trophic factors originate, and it does not seem to me that you have electrical activity in them where sodium channels may be involved.
Let us now look at the daunting list of chemical "substances" that have been used in research laboratories to counteract Nav1.7 sodium channels:
1. The benzazepinone65
2. Diaminotriazine. Bregman et al.66 recognized a type of diaminotriazine as a potent inhibitor of a Nav1.7 sodium channel subtype identified as hNav1.7.
3. The spiroxidol XEN90767
4. The pyrrolo-benzo-1,4-diazine as an inhibitor of Nav1.768
5. The indazole69
6. The shRNA70
7. JNJ63955918. On the assumption that inhibition of Nav1.7 sodium channels is responsible for pain insensitivity, starting with the OroTX-II peptide from tarantula venom, Flinspach et al,71 extracted a compound named JNJ63955918 that can pharmacologically induce pain insensitivity.
8. Chlorpromazine. Lee and Cll. 72 pointed out from their study that chlorpromazine blocks Nav.7 sodium channels: in fact, I have not found any other reports in the literature on this.
9. Jingzhaotoxin-34 (JZTX-34). Zeng et al73 claimed that under experimental conditions JZTX-34 which is a selective blocker of Nav1.7 sodium channels, at a dose of 2 mg/Kg has the same antalgic efficacy as 5 mg/Kg morphine.
10. The rSVmab 74
11. The acylsulfonamide75-78
12. The μ-theraphotoxin-Pn3a79
13. The DA-0218 (the unpronounceable: 9-(3-(1-benzyl-1H-indol-3-yl)-3-(3-phenoxyphenyl)-N-(2-(pyrrolidin-1-yl)ethyl)propanamide)80
14. Cytosolic collapsin response mediator protein (CRPM) which would be a regulator of Nav1.7 sodium channels effective in "chronic" but not "physiological" neuropathic pain81-82
15. The proTxII83-84
16. Propanamide85
17. Il-10. This product, as a Nav1.7 sodium channel inhibitor would reduce root pain in the rat 86
18. The tetrodotoxin87-88
19. The Nav1.7 inhibitor derived from ralphinamide and called QLS-8185
20. Amitriptyline for topical application as a potent inhibitor of Nav1.7, Nav1.8 and Nav1.9 sodium channels89
21. The ST-2530 or saxitoxin90
22. To add a note of folklore, Allium macrostemon Bunge was also considered, which is an edible herb used in traditional Chinese medicine to treat pain because it would block Nav1.7 sodium channels91
Counteracting NGF
Discovered in the 1950s by Italian researcher Rita Levi-Montalcini, who was awarded the Nobel Prize thanks to it, NGF is a protein that regulates the growth of sensory and sympathetic neurons during development and acts by binding to a specific receptor, which is tropomyosin kinase (TrK) present in the dorsal root ganglion and (presumably) in all sensory nerve fibers.
At first it was hoped that NGF might exert neurotrophic functions useful for nerve regeneration, but it was unsuccessful with this purpose. Almost ironically, later, having observed that NGF increases in inflammation because it is released by mast cells, macrophages, and lymphocytes, it was hypothesized that, because of its neurotrophic power, it might contribute to the production of hyperalgesia and allodynia in the tissues site of inflammation through sensitization of nociceptors. It was therefore considered a potency responsible for the persistence of pain and (obviously) its "chronicity".92 Along this line of thought, a monoclonal antibody (IgG) directed against the effect of NGF was prepared: Tanezumab, which acts by blocking the binding of NGF to its TrK receptor. What is hoped to be achieved with Tanezumab is the reduction of nociceptor excitability, i.e., what had been achieved until now with NSAIDs but with the difference that NSAIDs act only when nociceptor excitability is increased by prostaglandins and not when it depends, for example, on neurogenic inflammation that sustains neuroaptic pain from persistent nociceptor hyperexcitability. Thus, Tanezumab should find indication in tissue pain, neuropathic pain from persistent nociceptor hyperexcitability, and CRPS-I.
It should be considered, however, that the reduction in nociceptor excitability is achieved by Tanezumab by virtue of NGF antagonism and thus a neurotoxic effect: given this, we have to ask whether really the nerve damage we intend to produce with Tanezumab can be selectively limited to nociceptors without, as one may fear, involving axons or pyrenophores. Perhaps it is possible that the dosage of Tanezumab could be precisely calibrated to achieve this selectivity, but there is no getting away from the question of whether this is really possible.
A question arises at this point: can Tanezumab be considered a pain threshold elevator drug? The hypothetical drugs that counteract Nav1.7 sodium channels increase pain threshold because they counteract Nav1.7 sodium channels in nociceptors, and as an NGF inhibitor, Tanezumab "damages" nociceptors by reducing their excitability: therefore, Tanezumab can actually be considered a drug to elevate pain threshold.
It is evident that a drug with these characteristics may have great therapeutic potential in many clinical situations concerning tissue pain and some neuropathic pain. Having identified its potential as an analgesic, pre-clinical trials of Tanezumab began in the early 2000s for the control of pain related to osteoarthritis (especially of the hip and knee) and temporarily suspended in 2010 due to the reported occurrence of osteonecrosis in treated subjects. The "misfortune" of Tanezumab does not end there because, resumed worldwide experimentation, in 2021 a preparation of Tanezumab was proposed, but the European Medicines Agency denied the authorization.
Note that as side effects of Tanezumab are reported osteonecrosis, paresthesias, and "peripheral neuropathy" which paradoxically are among its therapeutic indications. Regarding osteonecrosis, one might think that the abolition of pain allows overuse of the joint involved in osteoarthritis. Perhaps NSAIDs do not produce osteonecrosis because they do not remove enough pain while Tanezumab produces it because it is more effective. This, on the one hand, confirms its antalgic efficacy and, on the other hand, because of the very narrow therapeutic range, implies the need not only for adequate dosing but also, and more importantly, the absolute necessity of its "controlled" use. In other words, one has to assess when and to what extent it "suits" to reduce nerve trophism. Keep in mind that according to a study,93 at least in monkeys, it appears that even at high doses Tanezumab has no neurotoxic effects when administered at 8-week intervals for 6 months but, despite these promising findings, we still do not know how long and in what doses it can be administered in humans with an acceptable margin of safety.
It is possible that Tanezumab will give birth to a new generation of analgesics 94,95 but, also because of its practicality of use, considering that it can be administered subcutaneously and has a long plasma half-life (about 3 weeks), it is easy to imagine that, caught up in the excitement, people will end up using it for all kinds of acute and, above all, "chronic" pain. Already it has been proposed not only for the treatment of coxarthrosis pain at a dose of 2.5-10 mg/8 weeks96,97 and bone metastasis pain,98 but also in low back pain99,100 and post-herpetic neuralgia.101 Three years ago,102 I wrote: "...one must hope that Tanezumab, on the one hand, is not one of the usual products that are as safe and harmless as they are useful only from a commercial point of view and, on the other hand, that it is really effective and sufficiently manageable in clinical practice." Since then, countless other studies on Tanezumab have appeared, but they have not led to any definitive conclusions.
As far as indications are concerned, peripheral neuropathy and post-herpetic neuralgia have ceased to be discussed and are considered exclusively osteoarthritis of the hip and knee and in some cases unspecified chronic low back pain,103,104
Problems not yet fully resolved concern the route of administration and dosing of Tanezumab. Regarding the first point, most studies propose subcutaneous administration at a dosage ranging from 2.5 to 10 mg every 8 weeks.105 For dosing, at first, Schnitzer et al,106 proposed the administration of 2.5 mg the first time and 5 mg after 8 weeks, but in a later publication the same Authors pointed out that the antalgic effect with the 5 mg dosage was only slightly higher than that obtained with 2.5 mg.107 Others, by contrast, proposed 5-10 mg subcutaneously every 8 weeks108 and Song and Lee109 argued that the optimal dosage is 5 mg/8 weeks.
As an alternative to subcutaneous and preferable to it, some authors proposed intravenous administration of Tanezumab. Walicke et al.110 proposed intravenous administration of 0.7 mg/Kg body weight for the treatment of knee osteoarthritis pain, and according to Cai et al.111 the best results with Tanezumab would be obtained with 10 mg intravenously and the worst with 2.5 mg subcutaneously. According to Markman et al,112 for the treatment of chronic low back pain, Tanezumab should be administered by the venous route at a dose of 2.5 or even 20 mg/8 weeks (in their experience the treatment lasted for 56 weeks).
It should be noted that, regardless of the route of administration, all authors agree in the 8-week interval between Tanezumab administrations (...so either the drug has a very long half-life or the preparation is retard).
Virtually everyone agrees on the antalgic efficacy of Tanezumab, which would give excellent antalgic results as early as the first week of treatment .112. Paradoxically, it would seem that the efficacy is even exaggerated since almost everyone reports the possilbility of aggravation of joint damage in patients with osteoarthritis of the hip or knee, 106,108,113-116 even though according to the reassuring observation of Zhang et al,117 such damage would occur only in a limited number of cases. Of the same opinion are Berenbaum et al,118 when they claim that paresthesias are frequent but aggravation of osteoarthritis is infrequent at the (recommended) dose of 2.5 mg/8 weeks (at least in the first 24 weeks). Ultimately, apart from the aggravation of osteoarthritis, the other side effects would be minimal,119 consisting of what in various studies are fumily referred to as "abnormal peripheral sensations" and which I believe should be understood as paresthesias.
Almost as a curiosity, the report of Konno et al,120 who claim that in "chronic low back pain," adverse effects (the usual "abnormal peripheral sensations") would affect 63% of patients given Tanezumab at a dose of 5 mg and 54% of those given 10 mg and also joint damage would be greater in those receiving 5 mg than in those receiving 10 mg.
Conclusion
I think it should be made clear that Nav1.7 sodium channel inhibitors and Tanezumab should by no means be equated with "analgesics": in clinical practice, they do not replace analgesics. What would be achieved with them is not simply the control of pain but even the elimination of the possibility of having pain, and the achievement of this mirage, which could be considered as one of the greatest achievements of modern man, in one respect can even be dangerous because, by exaggerating, one risks giving up the useful protective function of pain. In fact, this reasoning is the result of a misunderstanding that stems from the fact that what we should aim at is not the elimination of the possibility of feeling pain as a warning signal but the elimination of pain as a symptom after it has been used to pose the diagnosis of the disease that caused it and especially when it is disease per se. Of course there are nuances to consider in this reasoning. For example, there is no doubt that pain that prompts a reduction in the load on the knee joint in gonarthrosis serves to limit the aggravation of joint damage: however, after pain has signaled that pathology, one must provide for it by treating it appropriately, and it is unethical to rely on pain to contain its aggravation. In other words, the dangerousness of therapeutic elevation of the pain threshold can only occur if the achievement of this result corresponds to the folly of foregoing causal therapy when it is possible.
As a curiosity it should be pointed out that compared to the genome of modern humans, in Neanderthals there would have been three variants in the protein encoding Nav1.7 capable of resulting in their reduced inactivation so that peripheral nerves would have been more sensitive to nociceptive stimuli: in other words, Neanderthals felt more pain than modern humans and the above variants would be present in 4/1000 of present-day Britons.121 As ill-thinkers, it comes to consider that if it is true that Neanderthals felt pain more than modern humans the protective function of pain can be put into dicussion because Neanderthals who felt more pain have become extinct and modern humans who feel less pain have survived!
About 2 years ago, Tanezumab was proposed as a remedy for pain related to osteoarthritis, bone tumors, interstitial cystitis, and chronic low back pain...that is, as an alternative to traditional NSAID and opioid therapies: it should come as no surprise that on these premises on September 16, 2021, the European Medicines Agency refused marketing authorization for it.
Although commercially it seems convenient to extend the use of Tanezumab to clinical indications as frequent as generic as chronic low back pain, following this policy the drug will never be approved: in fact, its indications are not the suppression of pain at any cost but the elevation of the pain threshold in situations where it plays a relevant pathogenic role. Until its therapeutic indications are clarified, it is very unlikely that we will have this very interesting pharmacological device.
Tanezumab and the hypothetical Nav1.7 sodium channel inhibitors should by no means be considered as therapeutic alternatives but as options to be employed in particular and well-defined clinical situations, i.e., where the reduced pain threshold plays a particularly relevant pathogenetic role or even is the main pathogenetic element as in the case of Migraine.
These considerations lead to the determination that these drugs, far from being entrusted to generic use, should be managed with the utmost expertise. Thus, let us see when they should be used and when they should be preferred over common analgesics and infiltrative and surgical analgesic procedures.
Theoretically, Nav1.7 sodium channel inhibitors and Tanezumab should be used when a lower than normal pain threshold is found, but in practice this verification is almost impossible. Ultimately, since we can place little reliance on objective findings, the only way to decide whether or not to use Nav1. 7 sodium channel inhibitors and Tanezumab will have to be indirect clinical assessments, i.e., pathogenetic diagnosis of pain, considering them indicated:
1) in conditions where we believe that the main pathogenetic mechanism is reduced pain threshold, i.e., Migraine, Musculotensive Headache, Neuropathic Pain from Small Fiber Pathology, Burning Mouth Syndrome, Trigeminal Neuropathy, Phantom Limb Pain, Irritable Bowel Syndrome, Pain-prone Patient Status, Pain from Systemic Degenerative Disease in the Elderly, and... the much-fashionable Fibromyalgia;
1) in conditions where we believe that the main pathogenetic mechanism is reduced pain threshold, i.e., Migraine, Musculotensive Headache, Neuropathic Pain from Small Fiber Pathology, Burning Mouth Syndrome, Trigeminal Neuropathy, Phantom Limb Pain, Irritable Bowel Syndrome, Pain-prone Patient Status, Pain from Systemic Degenerative Disease in the Elderly, and... the much-fashionable Fibromyalgia;
2) in diseases where the pain threshold can be considered to condition the severity of symptomatology or even its onset;
3) when pain is no longer useful for diagnostic purposes;
4) when pain is due to a disease that cannot be cured such as cancer.
Conflict of interests
The authors declares no conflict of interests
Open Access-license (CC BY-NC 4.0)
Published
27th December 2023
Bibliografia
1) O'Driscoll S.L., Jayson M.I. Pain threshold analysis in patients with osteoarthrosis of hip. Br Med J., 3 (1974) 714-5
2) Imamura M. et al. Changes in pressure pain threshold in patients with chronic nonspecific low back pain. Spine (Phila Pa 1976), 38 (2013) 2098-107
3) Cygańska A.K. et al. Pain threshold in selected trigger points of superficial muscles of the back in young adults. PeerJ, 10 (2022) e12780
4) Eserdag S., Sevinc T., Tarlacı S. Do women with vaginismus have a lower threshold of pain? Eur J Obstet Gynecol Reprod Biol., 258 (2021) 189-192
5) Orlandini G. La semeiotica del dolore: dai presupposti teorici alla pratica clinica. Manuale d’uso pluridisciplinare. Seconda Edizione. Delfino Ed., Roma 2014
6) Incel N.A. et al. Pain pressure threshold values in ankylosing spondylitis. Rheumatol Int., 22 (2002) 148-50
7) Marques A.P. Quantifying pain threshold and quality of life of fibromyalgia patients. Clin Rheumatol., 24 (2005) 266-71
8) Gomes M.B. et al. Palpation and pressure pain threshold: reliability and validity in patients with temporomandibular disorders. Cranio, 26 (2008) 202-10.
9) Sayed-Noor A.S. et al. Pressure-pain threshold algometric measurement in patients with greater trochanteric pain after total hip arthroplasty. Clin J Pain, 24 (2008) 232-6
10) Terzi H., Terzi R., Kale A. The relationship between fibromyalgia and pressure pain threshold in patients with dyspareunia. Pain Res Manag., 20 (2015) 137-40.
11) Cheatham S.W et al. Concurrent validation of a pressure pain threshold scale for individuals with myofascial pain syndrome and fibromyalgia. J Man Manip Ther., 26 (2018) 25-35
12) Kelly de Oliveira A. et al. Reliability of pressure pain threshold on myofascial trigger points in the trapezius muscle of women with chronic neck pain. Rev Assoc Med Bras., 67 (2021) 708-712
13) Zicarelli C.A.M. et al. Reliability of pressure pain threshold to discriminate individuals with neck and low back pain. J Back Musculoskelet Rehabil., 34 (2021) 363-370.
14) Christidis N., Kopp S., Ernberg M. The effect on mechanical pain threshold over human muscles by oral administration of granisetron and diclofenac-sodium. Pain, 113 (2005) 265-270
15) Baek S.W., Erdek M.A. Time-dependent change in pain threshold following neurolytic celiac plexus block. Pain Manag., 9 (2019) 543-550
16) De Oliveira Gomes A. et al. Influence of different frequencies of transcutaneous electrical nerve stimulation on the threshold and pain intensity in young subjects. Einstein (Sao Paulo), 12 (2014) 318-22.17) Telles J.D et al. Transcutaneous electrical nerve stimulation and cervical joint manipulation on pressure pain threshold. Pain Manag., 8 (2018) 263-269.
18) Honoré M. et al. The regional effect of spinal manipulation on the pressure pain threshold in asymptomatic subjects: a systematic literature review. Chiropr Man Therap., 26 (2018) 11.
19) IASP (Subcommittee on Taxonomy) Classification of chronic pain: description of chronic pain syndromes and definitions of pain terms. IASP Press, Seattle 1994
20) Wilkerson J.L., Milligan E.D. The Central Role of Glia in Pathological Pain and the Potential of Targeting the Cannabinoid 2 Receptor for Pain Relief. ISRN Anesthesiol. 2011;2011(2011). pii: 593894
21) Hildebrand M.E., Smith P.L., Bladen C., Eduljee C., Xie J.Y., Chen L., Fee-Maki M., Doering C.J., Mezeyova J., Zhu Y., Belardetti F., Pajouhesh H., Parker D., Arneric S.P., Parmar M., Porreca F., Tringham E., Zamponi G.W., Snutch T.P. A novel slow-inactivation-specific ion channel modulator attenuates neuropathic pain. Pain, 152 (2011) 833-843
22) Orlandini G. Manuale della visita algologica e della formulazione della diagnosi. Delfino Ed., Roma 2022
23) Orlandini G. La decisione terapeutica nella medicina del dolore: dalla diagnosi patogenetica alla scelta motivata della terapia. Delfino Ed., Roma 2020
24) Kwon M., Jung Y., Cha M., Lee B.H. Inhibition of the Nav1.7 Channel in the Trigeminal Ganglion Relieves Pulpitis Inflammatory Pain. Front Pharmacol., 12 (2021)
25) Fischer TZ, Waxman S.G. Familial pain syndromes from mutations of the NaV1.7 sodium channel. Ann N Y Acad Sci., 1184 (2010) 196-207
26) Liu C., Cao J., Ren X., Zang W. Nav1.7 protein and mRNA expression in the dorsal root ganglia of rats with chronic neuropathic pain. Neural Regen Res., 7 (2012) 1540-4
27) Tian J.J. et al. Upregulation of Nav1.7 by endogenous hydrogen sulfide contributes to maintenance of neuropathic pain. Int J Mol Med., 46 (2020) 782-794
28) Estacion M. et al. Intra- and interfamily phenotypic diversity in pain syndromes associated with a gain-of-function variant of NaV1.7. Mol Pain, 7 (2011) 92
29) Themistocleous A.C. et al. The clinical approach to small fibre neuropathy and painful channelopathy. Pract Neurol., 14 (2014) 368-79
30) Doppler K., Sommer C. Neuropathic pain associated with Nav1.7 mutations: clinical picture and treatment. Nervenarzt., 84 (2013) 1428-35
31) Blass B.E. Acyl Sulfonamides NaV1.7 Blockers Useful for the Treatment of Pain. ACS Med Chem Lett. 9 (2018) 161–162.
32) De Rooij A.M., Gosso M.F., Alsina-Sanchis E., Marinus J., Van Hilten J.J., Van den Maagdenberg A.M.J.M. No mutations in the voltage-gated NaV1.7 sodium channel alpha1 subunit gene SCN9A in familial complex regional pain sindrome. Eur J Neurol., 17 (2010) 808-14.
33) Long Zhang X. et al. Foxo 1 selectively regulates static mechanical pain by interacting with Nav1.7. Pain, 162 (2021) 490-502.
34) Tilley P., Bisset L. The Reliability and Validity of Using Ice to Measure Cold Pain Threshold. Biomed Res Int., (2017) 7640649.
35) Buchanan H.M., Midgley J.A. Evaluation of pain threshold using a simple pressure algometer. Clin Rheumatol., 6 (1987) 510-7.
36) Hogeweg J.A. et al. Algometry. Measuring pain threshold, method and characteristics in healthy subjects. Scand J Rehabil Med., 24 (1992) 99-103.
37) Intonaci F. et al. Pain threshold in humans. A study with the pressure algometer. Funct Neurol., 7 (1992) 283-8.
38) Melia M. et al. Measuring mechanical pain: the refinement and standardization of pressure pain threshold measurements. Behav Res Methods., 47 (2015) 216-27.
39) Mikkelsson et al. Muscle and bone pressure pain threshold and pain tolerance in fibromyalgia patients and controls. Arch Phys Med Rehabil., 73 (1992) 814-8.
40) Montenegro M.L.L.S. et al. Pain pressure threshold algometry of the abdominal wall in healthy women. Braz J Med Biol Res., 45 (2012) 578-82.
41) Maresca M, Faccani G. The measurement of pain threshold in man by means of electrical stimuli. A critical appraisal. J Neurosurg Sci., 27 (1983) 83-93.
42) Chen R., Coppes O.J.M., Urman R.D. Receptor and Molecular Targets for the Development of Novel Opioid and Non-Opioid Analgesic Therapies. Pain Physician, 24 (2021) 153-163.
43) Nassar M.A. et al. Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proc Natl Acad Sci U S A, 101 (2004) 12706-11
44) Strickland I.T., Martindale J.C., Woodhams P.L., Reeve A.J., Chessell I.P., McQueen D.S. Changes in the expression of NaV1.7, NaV1.8 and NaV1.9 in a distinct population of dorsal root ganglia innervating the rat knee joint in a model of chronic inflammatory joint pain. Eur J Pain, 12 (2008) 564-72
45) Kanellopoulos A.H., Matsuyama A. Voltage-gated sodium channels and pain-related disorders. Clin Sci (Lond), 130 (2016) 2257-2265
46) Foadi N. Modulation of sodium channels as pharmacological tool for pain therapy-highlights and gaps. Naunyn Schmiedebergs Arch Pharmacol., 391 (2018) 481-488
47) Abdel-Magid A.F. Nav1.7 Inhibitors: Potential Effective Therapy for the Treatment of Chronic Pain. ACS Med Chem Lett., 6 (2015) 956-7
48) Hao J.X., Stöhr T., Selve N., Wiesenfeld-Hallin Z., Xu X.J. Lacosamide, a new anti-epileptic, alleviates neuropathic pain-like behaviors in rat models of spinal cord or trigeminal nerve injury. Eur J Pharmacol., 553 (2006) 135-40
49) Stöhr T., Krause E., Selve N. Lacosamide displays potent antinociceptive effects in animal models for inflammatory pain. Eur J Pain, 10 (2006) 241-9
50) Beyreuther B.K., Geis C., Stöhr T., Sommer C. Antihyperalgesic efficacy of lacosamide in a rat model for muscle pain induced by TNF. Neuropharmacology, 52 (2007) 1312-7
51) Biton V. Lacosamide for the treatment of diabetic neuropathic pain. Expert Rev Neurother., 8 (2008) 1649-60.
52) Harris J.A., Murphy J.A. Lacosamide: an adjunctive agent for partial-onset seizures and potential therapy for neuropathic pain. Ann Pharmacother., 43 (2009) 1809-17.
53) McCleane G. Lacosamide for pain. Expert Opin Investig Drugs., 19 (2010) 1129-34.
54) Hearn L., Derry S., Moore R.A. Lacosamide for neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev. 2012.
55) Masrour S. Lacosamide for refractory trigeminal neuralgia and other facial pain. Case report. Headache, 62 (2022) 1227-1230.
56) Alcántara Montero A. Off-label use of lacosamide, an alternative for the treatment of neuropathic pain. Headache., 62 (2022) 1239-1240.
57) Goldberg Y.P. et al. Loss-of-function mutations in the Nav1.7 gene underlie congenital indifference to pain in multiple human populations. Clin Genet., 71 (2007) 311-9.
58) King G.F., Vetter I. No gain, no pain: NaV1.7 as an analgesic target. ACS Chem Neurosi., 5 (2014) 749-51.
59) Hoffmann T, Sharon O., Wittmann J., Carr R.W., Vyshnevska A., Col R., Nassar M.A., Reeh P.W. NaV1.7 and pain: contribution of peripheral nerves. Pain, 159 (2018) 496-506.
60) Shields S.D. et al. Insensitivity to Pain upon Adult-Onset Deletion of Nav1.7 or Its Blockade with Selective Inhibitors. J Neurosi., 38 (2018) 10180-10201
61) Emery E.C., Luiz A.P., Wood J.N. Nav1.7 and other voltage-gated sodium channels as drug targets for pain relief. Expert Opin Ther Targets, 20 (2016) 975-83
62) Hoffmann T., Sharon O., Wittmann J., Carr R.W., Vyshnevska A., Col R., Nassar M.A., Reeh P.W. NaV1.7 and pain: contribution of peripheral nerves. Pain, 159 (2018) 496-506
63) Doppler K., Sommer C. Neuropathic pain associated with Nav1.7 mutations: clinical picture and treatment. Nervenarzt., 84 (2013) 1428-35.
64) Blass B.E. Acyl Sulfonamides NaV1.7 Blockers Useful for the Treatment of Pain. ACS Med Chem Lett. 9 (2018) 161–162.
65) Hoyt S.B. et al. Benzazepinone Nav1.7 blockers: potential treatments for neuropathic pain. Bioorg Med Chem Lett., 17 (2007) 6172-7.
66) Bregman H. et al. Identification of a potent, state-dependent inhibitor of Nav1.7 with oral efficacy in the formalin model of persistent pain. J Med Chem., 54 (2011) 4427-45.
67) Chowdhury S. et al. Discovery of XEN907, a spirooxindole blocker of NaV1.7 for the treatment of pain. Bioorg Med Chem Lett., 21 (2011) 3676-81.
68) Ho G.D. et al. Discovery of pyrrolo-benzo-1,4-diazines as potent Na(v)1.7 sodium channel blockers. Bioorg Med Chem Lett., 24 (2014) 4110-3.
69) Frost J.M. et al. Substituted Indazoles as Nav1.7 Blockers for the Treatment of Pain. J Med Chem., 59 (2016) 3373-91.
70) Cai W., Cao J., Ren X., Qiao L., Chen X., Li M., Zang W. shRNA mediated knockdown of Nav1.7 in rat dorsal root ganglion attenuates pain following burn injury. BMC Anesthesiol., 16 (2016) 59
71) Flinspach M. et al. Insensitivity to pain induced by a potent selective closed-state Nav1.7 inhibitor. Sci Rep., 7 (2017) 39662
72) Lee S.J., Kim D.H., Hahn S.J., Waxman S.G., Choi J.S. Mechanism of inhibition by chlorpromazine of the human pain threshold sodium channel, Nav1.7. Neurosci Lett., 639 (2017) 1-7
73) Zeng X. et al. Selective Closed-State Nav1.7 Blocker JZTX-34 Exhibits Analgesic Effects against Pain. Toxins (Basel), 10 (2018) 64
74) Sangsu Bang B. et al. Differential Inhibition of Nav1.7 and Neuropathic Pain by Hybridoma-Produced and Recombinant Monoclonal Antibodies that Target Nav1.7 : Differential activities of Nav1.7-targeting monoclonal antibodies. Neurosci Bull., 34 (2018) 22-41.
75) Focken T. et al. Discovery of Aryl Sulfonamides as Isoform-Selective Inhibitors of NaV1.7 with Efficacy in Rodent Pain Models. ACS Med Chem Lett., 7 (2016) 277-82.
76) Blass B.E. Acyl Sulfonamides NaV1.7 Blockers Useful for the Treatment of Pain. ACS Med Chem Lett. 9 (2018) 161–162.
77) Shields S.D. et al. Insensitivity to Pain upon Adult-Onset Deletion of Nav1.7 or Its Blockade with Selective Inhibitors. J Neurosi., 38 (2018) 10180-10201.
78) Safina B.S. et al. Discovery of Acyl-sulfonamide Nav1.7 inhibirors GDC-9276 and GDC-0310. J.Med.Chem., 64 (2021) 2953-2966.
79) Mueller A. et al. Antiallodynic effects of the selective NaV1.7 inhibitor Pn3a in a mouse model of acute postsurgical pain: evidence for analgesic synergy with opioids and baclofen. Pain, 160 (2019) 1766-1780.
80) Chandra S., Wang Z., Tao X., Chen O., Luo X., Bortsov AV. Computer-aided Discovery of a New Nav1.7 Inhibitor for Treatment of Pain and Itch. Anesthesiology, 133 (2020) 611-627.
81) Moutal A. et al. Studies on CRMP2 SUMOylation-deficient transgenic mice identify sex-specific Nav1.7 regulation in the pathogenesis of chronic neuropathic pain. Pain, 161 (2020) 2629-2651.
82) Li J., Stratton H.J., Lorca S.A., Grace P.M., Khanna R. Small molecule targeting NaV1.7 via inhibition of the CRMP2-Ubc9 interaction reduces pain in chronic constriction injury (CCI) rats. Channels (Austin), 16 (2022) 1-8.
83) Abdel-Magid A.F. Nav1.7 Inhibitors: Potential Effective Therapy for the Treatment of Chronic Pain. ACS Med Chem Lett., 6 (2015) 956-7.
84) Kwon M., Jung Y., Cha M., Lee B.H. Inhibition of the Nav1.7 Channel in the Trigeminal Ganglion Relieves Pulpitis Inflammatory Pain. Front Pharmacol., 12 (2021).
85) Niu H. et al. Inhibition of Nav1.7 channel by a novel blocker QLS-81 for alleviation of neuropathic pain. Acta Pharmacol Sin., 42 (2021) 1235-1247.
86) Huang Y., Zhu L., Zhang W., Tang Q., Zhong Y. IL-10 alleviates radicular pain by inhibiting TNF-α/p65 dependent Nav1.7 up-regulation in DRG neurons of rats. Brain Res., 1791 (2022) 147997.
87) Salas M.M. et al. Tetrodotoxin suppresses thermal hyperalgesia and mechanical allodynia in a rat full thickness thermal injury pain model. Neurosci Lett., 607 (2015) 108-113.
88) Mattei C. Tetrodotoxin, a Candidate Drug for Nav1.1-Induced Mechanical Pain? Mar Drugs, 16 (2018) 72.
89) Genevois A.L. et al. Analgesic Effects of Topical Amitriptyline in Patients With Chemotherapy-Induced Peripheral Neuropathy: Mechanistic Insights From Studies in Mice. J Pain, 22 (2021) 440-453.
90) Beckley J.T. et al. Antinociceptive properties of an isoform-selective inhibitor of Nav1.7 derived from saxitoxin in mouse models of pain. Pain, 162 (2021) 1250-1261
91) Yang X. et al. Allium macrostemon Bunge. exerts analgesic activity by inhibiting NaV1.7 channel. J Ethnopharmacol., 281 (2021) 114495
92) Patel M.K., Kaye A.D., Urman R.D. Tanezumab: Therapy targeting nerve growth factor in pain pathogenesis. J Anaesthesiol Clin Pharmacol., 34 (2018) 111-116
93) Belanger P., Butler P., Butt M., Bhatt S., Foote S., Shelton D., Evans M., Arends R., Hurst S., Okerberg C., Cummings T., Potter D., Steidl-Nichols J., Zorbas M. From the Cover: Evaluation of the Effects of Tanezumab, a Monoclonal Antibody Against Nerve Growth Factor, on the Sympathetic Nervous System in Adult Cynomolgus Monkeys (Macaca fascicularis): A Stereologic, Histomorphologic, and Cardiofunctional Assessment. Toxicol Sci., 158 (2017) 319-333
94) Brown M.T., Herrmann D.N., Goldstein M., Burr A.M., Smith M.D., West C.R., Verburg K.M., Dyck P.J. Nerve safety of tanezumab, a nerve growth factor inhibitor for pain treatment. J Neurol Sci., 345 (2014) 139-147
95) Nair A.S. Tanezumab: Finally a Monoclonal Antibody for Pain Relief Indian J Palliat Care. 24 (2018) 384–385.
96) Birbara C., Dabezies E.J. Jr., Burr A.M., Fountaine R.J., Smith M.D., Brown M.T., West C.R., Arends R.H., Verburg K.M. Safety and efficacy of subcutaneous tanezumab in patients with knee or hip osteoarthritis. J Pain Res., 11 (2018) 151-164.
97) Ekman E.F., Gimbel J.S., Bello A.E., Smith M.D., Keller D.S., Annis K.M., Brown M.T., West C.R., Verburg K.M. Efficacy and safety of intravenous tanezumab for the symptomatic treatment of osteoarthritis: 2 randomized controlled trials versus naproxen. J Rheumatol. 41 (2014) 2249-2259
98) Sopata M., Katz N., Carey W., Smith M.D., Keller D., Verburg K.M., West C.R., Wolfram G., Brown M.T. Efficacy and safety of tanezumab in the treatment of pain from bone metastases. Pain, 156 (2015) 1703-1713
99) Jayabalan P., Schnitzer T.J. Tanezumab in the treatment of chronic musculoskeletal conditions. Expert Opin Biol Ther., 17 (2017) 245-254.
100) Webb M.P., Helander E.M., Menard B.L., Urman R.D., Kaye A.D. Tanezumab: a selective humanized mAb for chronic lower back pain. Ther Clin Risk Manag., 14 (2018) 361-367.
101) Bramson C., Herrmann D.N., Carey W., Keller D., Brown M.T., West C.R., Verburg K.M., Dyck P.J. Exploring the role of tanezumab as a novel treatment for the relief of neuropathic pain. Pain Med., 16 (2015) 1163-1176.
102) Orlandini G. La decisione terapeutica nella medicina del dolore: dalla diagnosi patogenetica alla scelta motivata della terapia. Delfino Ed., Roma 2020.
103) Markman J.D. et al. Tanezumab for chronic low back pain: a randomized, double-blind, placebo- and active-controlled, phase 3 study of efficacy and safety. Pain, 161 (2020) 2068-2078.
104) Konno S.I., Nikaido T., Markman J.D., Ohta M., Machida T., Isogawa N., Yoshimatsu H., Viktrup L., Brown M.T., West C.R., Verburg K.M. Tanezumab for chronic low back pain: a long-term, randomized, celecoxib-controlled Japanese Phase III safety study. Pain Manag., 12 (2022) 323-335
105) Birbara C., Dabezies E.J. Jr., Burr A.M., Fountaine R.J., Smith M.D., Brown M.T., West C.R., Arends R.H., Verburg K.M. Safety and efficacy of subcutaneous tanezumab in patients with knee or hip osteoarthritis. J Pain Res., 11 (2018) 151-164.
106) Schnitzer T.J. et al. Effect of Tanezumab on Joint Pain, Physical Function, and Patient Global Assessment of Osteoarthritis Among Patients With Osteoarthritis of the Hip or Knee: A Randomized Clinical Trial. JAMA, 322 (2019) 37-48.
107) Schnitzer T.J. et al. Onset and maintenance of efficacy of subcutaneous tanezumab in patients with moderate to severe osteoarthritis of the knee or hip: A 16-week dose-titration study. Semin Arthritis Rheum., 50 (2020) 387-393.
108) Markman J.D. et al. Tanezumab for chronic low back pain: a randomized, double-blind, placebo- and active-controlled, phase 3 study of efficacy and safety. Pain, 161 (2020) 2068-2078
109) Song G.G., Lee Y.H. Relative efficacy and tolerability of 2.5, 5, and 10 mg tanezumab for the treatment of osteoarthritis: A Bayesian network meta-analysis of randomized controlled trials based on patient withdrawal. Int J Clin Pharmacol Ther. 59 (2021) 147-155
110) Walicke P.A. et al. First-in-human randomized clinical trials of the safety and efficacy of tanezumab for treatment of chronic knee osteoarthritis pain or acute bunionectomy pain. Pain Rep., 3 (2018) 653.
111) Cai Y.Z., Nie L.Y., Ruan J.Q., Zhao K. Effectiveness of Various Dosages and Administration Methods of Tanezumab for the Treatment of Pain in Knee and Hip Osteoarthritis: a Network Meta-Analysis. Clin Ther., 43 (2021) 2116-2126
112) Markman J.D., Schnitzer T.J., Perrot S., Beydoun S.R., Ohtori S., Viktrup L., Yang R., Bramson C., West C.R., Verburg K.M. Clinical Meaningfulness of Response to Tanezumab in Patients with Chronic Low Back Pain: Analysis From a 56-Week, Randomized, Placebo- and Tramadol-Controlled, Phase 3 Trial. Pain Ther., 11 (2022) 1267-1285
113) Berenbaum F, Langford R., Perrot S., Miki K., Blanco F.J., Yamabe T., Isogawa N., Junor R., Carey W., Viktrup L., West C.R., Brown M.T., Verburg K.M. Subcutaneous tanezumab for osteoarthritis: Is the early improvement in pain and function meaningful and sustained? Eur J Pain, 25 (2021) 1525-1539
114) Berenbaum F. et al. Subcutaneous tanezumab for osteoarthritis of the hip or knee: efficacy and safety results from a 24-week randomised phase III study with a 24-week follow-up period. Ann Rheum Dis., 79 (2020) 800-810
115) Yu Y., Lu S.T., Sun J.P., Zhou W. Safety of Low-Dose Tanezumab in the Treatment of Hip or Knee Osteoarthritis: A Systemic Review and Meta-analysis of Randomized Phase III Clinical Trials. Pain Med., 22 (2021) 585-595
116) Miki K., Ohta M., Abe M., Yoshimatsu H., Fujii K., Ebata N., West C.R., Brown M.T., Pixton G., Isogawa N. Efficacy, General Safety, and Joint Safety of Tanezumab in Japanese Patients with Osteoarthritis: Subgroup Analyses from Two Randomized, Phase 3 Studies. Pain Ther., 11 (2022) 827-844
117) Zhang B., Tian X., Qu Z., Liu J., Yang L. Relative Efficacy and Safety of Tanezumab for Osteoarthritis: A Systematic Review and Meta-analysis of Randomized-Controlled Trials. Clin J Pain, 37 (2021) 914-924
118) Berenbaum F., Schnitzer T.J., Kivitz A.J., Viktrup L., Hickman A., Pixton G., Brown M.T., Davignon I., West C.R. General Safety and Tolerability of Subcutaneous Tanezumab for Osteoarthritis: A Pooled Analysis of Three Randomized, Placebo-Controlled Trials. Arthritis Care Res (Hoboken)., 74 (2022) 918-928
119) Fan Z.R. et al. Efficacy and safety of tanezumab administered as a fixed dosing regimen in patients with knee or hip osteoarthritis: a meta-analysis of randomized controlled phase III trias. Clin Rheumatol., 40 (2021) 2155-2165.
120) Konno S.I., Nikaido T., Markman J.D., Ohta M., Machida T., Isogawa N., Yoshimatsu H., Viktrup L., Brown M.T., West C.R., Verburg K.M. Tanezumab for chronic low back pain: a long-term, randomized, celecoxib-controlled Japanese Phase III safety study. Pain Manag., 12 (2022) 323-335.
121) Zeberg H, Dannemann M., Sahlholm K., Tsuo K., Maricic T., Wiebe V., Hevers W., Robinson H.P.C., Kelso J., Pääbo S. A Neanderthal Sodium Channel Increases Pain Sensitivity in Present-Day Humans. Curr Biol., 30 (2020) 3465-3469.